D. Müller, R. Beckert*
Prof. Dr. R. Beckert
Institut fuer Organische Chemie und Makromolekulare Chemie
Friedrich-Schiller-Universiät-Jena
Humboldtstr. 10
07743 Jena
c6bera@rz.uni-jena.de
Einleitung:
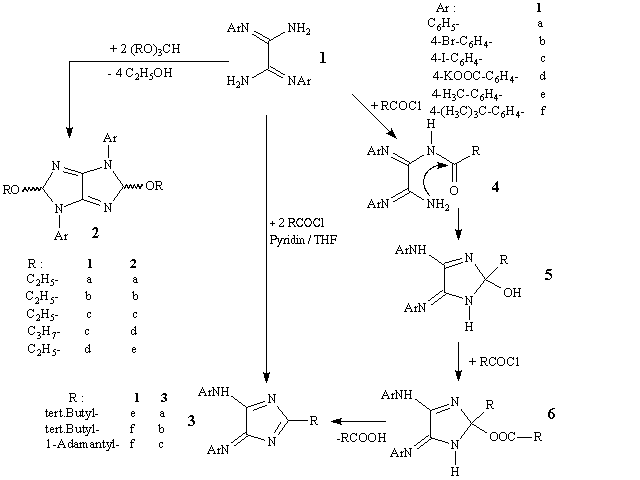
Kürzlich berichteten wir über einen einfachen Zugang zu N,N'-disubstituierten Oxalamidinen des Typs 1 und deren leichte Transformation in die entsprechenden 1,2,4,5-Tetrahydro-imidazo(4,5-d)imidazole 2, bzw. in die 4H-Imidazole des Typs 3 (D. Müller, R.Beckert, H. Görls; Synthesis; 601-606; (2001)). Über die Synthese von alkylsubstituierten 4H-Imidazolen 3, ausgehend von den Amidinen 1, sowie dem Verhalten von 1 gegenüber aliphatischen Carbonsäurechloriden mit alpha-ständigem Wasserstoff wurde in (D. Müller, R.Beckert, J.Weston, H. Görls, W. Günther, M. Friedrich; Eur. J. Org. Chem.; in press; (2001)) publiziert. Im folgenden sollen sowohl einige weitere Derivate der Imidazo(4,5-d)imidazole 2 vorgestellt und auch auf die Darstellung alkylsubstituierter 4H-Imidazole des Typs 3 näher eingegangen werden.
Hauptteil:
In die Synthese der Tetrahydro-imidazo(4,5-d)imidazole 2 gelang es weitere aromatische Reste wie z.B: Phenyl-, 4-Bromphenyl- und 4-Iodphenyl- zu involvieren. Auch diese Verbindungen (2a-d) fallen als Gemisch von Diastereomeren an und zeigen in ihren NMR-Spektren doppelte Signalsätze. Ausgehend vom Oxalamidin 1d konnte erstmals ein wasserlösliches Derivat 2e erhalten werden. Eine besondere Eigenschaft der Oxalamidine 1 sind die Cyclisierungsreaktionen mit Acylchloriden unter Bildung von 4H-Imidazolen bei Anwesenheit von n-Butyllithium (D. Müller, R.Beckert, H. Görls; Synthesis; 601-606; (2001)). Als Hilfsbase für diese Reaktion konnte auch Pyridin verwendet werden (D. Müller, R.Beckert, J.Weston, H. Görls, W. Günther, M. Friedrich; Eur. J. Org. Chem.; in press; (2001)). Hierbei zeigte sich jedoch, dass die Abspaltung von Wasser aus dem Intermediat 5 behindert war und die Derivate 3 nur in geringen Ausbeuten gebildet wurden. Erst der Zusatz eines weiteren Äquivalents an Acylchlorid ermöglichte, nach einer intermediären O-Acylierung und anschließenden Carbonsäureabspaltung, eine vollständigere Umsetzung der Oxalamidine 1 zu den 4H-Imidazolen 3. Durch diese neue Synthese gelang es erstmals auf einfachem Wege, alkylsubstituierte 4H-Imidazole des Typs 3 darzustellen. Die Derivate 3a-c zeigen in ihren 13C-NMR-Spektren die für diese Verbindungen typische starke Tieffeldverschiebung des C2-Signals des Imidazolrings bei ca. 200 ppm.
Experimenteller Teil:
Alle verwendeten Chemikalien sind, falls nicht extra aufgeführt, kommerziell verfügbar (Firma Aldrich, Fluka, Merck, Lancaster) und wurden ohne weitere Reinigung eingesetzt. Lösungsmittel, wie z.B.: n-Heptan, THF, Toluen oder Chloroform, wurden mittels Standardtechniken vor der Verwendung absolutiert und gereinigt. Der Reaktionsverlauf wurde dünnschichtchromatographisch verfolgt, wobei Fertigplatten, beschichtet mit neutralem Aluminiumoxid und Fluoreszenzindikator (Polygram ALOX N/UV(254) der Firma Marchery-Nagel verwendet wurden. Als stationäre Phase bei der präparativen Säulenchromatographie diente neutrales Aluminiumoxid (Merck, Aluminiumoxid 90, aktiv-neutral, Aktivität V, Korngröße 0,063-0,2 mm, 70-230 mesh ASTM). Die als mobile Phase verwendeten Lösungsmittel sind bei den entsprechenden Synthesevorschriften aufgeführt. Die Schmelzpunkte wurden auf einem Mikroheiztisch Galen III nach Boetius der Firma Cambridge Instruments oder mittels Digital Melting Point Analyzer KSPS 1000 der Firma A. Krüss-Optronic bestimmt und sind unkorrigiert. Die Aufnahme der IR-Spektren erfolgte mit dem IR-Spektrometer FTS-25 der Firma Bio-Rad, die der UV-Vis-Spektren an einem Lambda-19-Spektrometer der Firma Perkin-Elmer. Für alle neuen Substanzen wurden Elementaranalysen mit dem CHN-Automaten CHNS-832 der Firma Leco angefertigt und sie bewegen sich innerhalb der gerätespezifischen Toleranzbereiche. Die Massenspektren wurden an einem Gerät der Marke Finnigan MAT SSQ 710, die hochaufgelösten Massenpektren mit einem Finnigan MAT 95 XL-Trap aufgenommen. Die 1H- bzw. 13C-NMR-Spektren wurden an einem AC-250 (250 MHz)- bzw. DRX-400 (400 MHz)-Spektrometer der Firma Bruker gemessen (NMR-Shift relativ zum verwendeten deuterierten Lösungsmittel).
Synthese der 2,5-Dialkoxy-1,4-diaryl-1,2,4,5-tetrahydro-imidazo(4,5-d)imidazole:
1 mmol des Oxalamidins 1 wird in 20 ml des entsprechenden Trialkoxyorthoformiats suspendiert und unter Argon 4-8 h auf 120 °C erwärmt. Nach vollständigem Umsatz (DC-Kontrolle: Toluol / Aceton 10:1) engt man die Reaktionslösung um die Hälfte ein und läßt bei 0 °C kristallisieren. Das Reaktionsprodukt wird abgesaugt, mit n-Heptan gewaschen und anschließend getrocknet.
2,5-Diethoxy-1,4-diphenyl-1,2,4,5-tetrahydro-imidazo[4,5-d]imidazol 2a:
Ausbeute: 52%, grau-weiße Kristalle, m.p.: 97°C.
MS(CI): 350(60)[M]; 305(15); 276(40); 175(95); 147(50); 119(100); 77(60); 29(30).
IR(ATR): 2978 (CH-aliph.); 2937; 2896; 1680 (C=N-); 1593; 1502; 1414; 1388; 1360; 1175; 1097; 1028; 886; 757; 732; 689; 657.
H-NMR(250 MHz, CDCl3): 1,22 (m, 6H, CH3-); 3,49 (br., m, 4H,-CH2-O-); 7,13 (m, ges.: 4H, 2H: C(asym)H / 2H: 4-CH-Phenyl.); 7,42 (t, a,b-Aromat, J=7,9 Hz, 3-CH-Phenyl.); 7,84 (t, a,b-Aromat, J= 8,1 Hz, 2-CH-Phenyl.).
C-NMR (100 MHz, CDCl3): 14,7 (CH3); 57,2 (-CH2-O); 57,4 (-CH2-O); 111,7 (C(asym)H); 112,8 (C(asym)H); 117,5/ 117,6 (2-C-Phenyl.); 124,0 (4-C-Phenyl.); 129,3 (3-C-Phenyl.); 137,2 (C (quart.), ipso-N).
2,5-Diethoxy-1,4-di-p-bromphenyl-1,2,4,5-tetrahydro-imidazo[4,5-d]imidazol 2b:
Ausbeute: 68%, grau-weiße Kristalle, m.p.: 240-242°C.
MS(CI): 509(100)[M+1]; 463(30); 312(10); 256(15); 154(10); 103(15).
IR(ATR): 3095 (CH-arom.); 2977 (CH-aliph.); 2940; 2898; 2865; 1666 (C=N-); 1588; 1492; 1420; 1405; 1350; 1178; 1090; 1040; 1028; 882; 825; 814; 681; 666.
H-NMR (400 MHz, CDCl3): 1,19 (m, 6H, CH3-); 3,49 (m, 4H, -CH2-O-); 7,01/7,05 (2*s, ges. 2H, C(asym)H); 7,52 (m, 4H, CH-arom.); 7,71 (m, 4H, CH-arom.).
C-NMR (100 MHz,CDCl3): 14,7/14,8 (CH3); 57,6 (-CH2-O); 57,7 (-CH2-O); 111,9 (C(asym)H); 113,0 (C(asym)H); 117,0 (C(quart.), ipso-Br); 119,1/119,2 (CH-arom.); 132,4 (CH-arom.); 136,2 (C(quart.), ipso-N); 157,6 (C(quart.)); 158,3 (C(quart.)).
2,5-Diethoxy-1,4-di-p-iodphenyl-1,2,4,5-tetrahydro-imidazo[4,5-d]imidazol 2c:
Ausbeute: 65%, grau-weiße Kristalle, m.p.: 206-209°C.
MS(CI): 603(100)[M+1]; 557(35); 476(42); 431(12); 301(15); 103(32).
IR(ATR): 3294; 3091 (CH-arom.); 2976 (CH-aliph.); 2929; 2892; 1682 (C=N-); 1584; 1491; 1422; 1400; 1098; 1042; 1026; 1002; 883; 817; 670.
H-NMR (400 MHz, CDCl3): 1,19 (m, 6H, CH3-); 3,48 (m, 4H, -CH2-O-); 7,00/7,04 (2*s, ges. 2H, C(asym)H); 7,65 (m, 8H, CH-arom.).
C-NMR (100 MHz, CDCl3): 14,6/14,7 (CH3); 57,5 (-CH2-O); 57,6 (-CH2-O); 87,5 (C(quart.), ipso-I); 111,7 ( C(asym)H); 112,8 ( C(asym)H); 119,3/119,4 (CH-arom.); 136,76/136,77 (C (quart.)); 138,18/138,19 (CH-arom.); 157,5 (C(quart.)); 158,1 (C(quart.)).
2,5-Dipropoxy-1,4-di-p-iodphenyl-1,2,4,5-tetrahydro-imidazo[4,5-d]imidazol 2d:
Ausbeute: 58%, grau-weiße Kristalle, m.p.: 173-176° C.
MS(CI): 631(100)[M+1]; 571(43); 504(40); 493(25); 445(15); 315(15); 273(17); 256(23); 154(17); 107(17).
IR(ATR): 3425; 3294; 3087 (CH-arom.); 2955 (CH-aliph.); 2925; 2885; 1665 (C=N-); 1583; 1488; 1401; 1090; 1039; 1001; 882; 815; 735; 666.
H-NMR (400 MHz,CDCl3): 0,88 (m, 6H, CH3); 1,59 (m, 4H, -CH2-); 3,35 (m, 4H, -CH2-O-); 7,02/7,05 (2*s, ges. 2H, C(asym)H); 7,65 (m, 8H, CH-arom.).
C-NMR (100 MHz, CDCl3): 10,5/10,6 (CH3); 22,3/22,4 (-CH2-); 63,4 (-CH2-O); 63,6 (-CH2-O); 87,5 (C(quart.), ipso-I); 111,8 ( C(asym)H); 112,9 (C(asym)H); 119,3/119,4 (CH-arom.); 136,8 (C(quart.)); 138,1/138,2 (CH-arom.); 138,5 (C(quart.)); 157,5 (C(quart.)); 158,1 (C(quart.)).
2,5-Diethoxy-1,4-di-p-carboxyphenyl-1,2,4,5-tetrahydro-imidazo[4,5-d]imidazol-di-Kalium-Salz 2e:
Das Oxalamidin 1d (1 mmol, 402 mg) wird zusammen mit 30 ml Triethylothoformiat 24 Stunden unter Rückfluß erhitzt. Das entstandene Salz wird abfiltriert, mit n-Heptan gewaschen und getrocknet.
Ausbeute: 61%, grau-weißes Pulver, m.p.: ab 366°C Zersetzung.
MS(DEI): Molpeak nicht detektiert 382(30); 337(10); 240(10); 218(40); 212(50); 165(60); 137(50); 120(100); 93(30); 65(25).
IR(ATR): 3095 (CH-arom.); 2977; 2940; 2898 (CH-aliph.); 1666 (C=O/C=N-); 1588 (C=C); 1492; 1420; 1405; 1350; 1178; 1090; 1040; 1028; 882; 825; 814; 681; 666.
H-NMR (400 MHz,D2O): 1,12 (t, J=7,0)/ 1,29(t, J=4,7) gesamt 6H, (CH3); 3,59 (q, J=7,1)/ 4,28 (q, J=7,1), gesamt 4H, (-CH2-O-); 7,02 (m, 6H, C(asym)H / CH-arom.); 7,83 (m, 4H, CH-arom.).
C-NMR (100 MHz, D2O): 13,5/14,0 (CH3); 61,1/62,1 (CH2); 121,5/122,0 (CH); 130,7(CH); 131,3/132,0 (C(quart.)); 152,6/153,0 (C(quart.)); 175,38/175,41(-COO(-)).
Vorschrift zur Synthese der alkylsubstituierten 4H-Imidazole des Typs 3
1 mmol des entsprechenden Oxalamidins 1 wird zusammen mit 1 ml trockenem Pyridin in 20 ml getrocknetem THF gelöst. Anschließend setzt man 2 mmol des korrespondierenden Acylchlorides zu und erhitzt eine Stunde unter Rückfluß. Die Reaktionslösung wird komplett eingeengt und das entstandene 4H-Imidazol 3 säulenchromatographisch gereinigt (Laufmittel: Toluol/Aceton 10:1).
2-(tert.)-Butyl-5-(4-methylphenylamino)-4-(4-methylphenylimino)-4H-imidazol 3a:
Ausbeute: 70 %, orange Kristalle, m.p.: 127-129°C.
MS(CI): 333(100)[M+1]; 317(25).
IR(ATR): 3352 (NH); 3032 (CH-arom.); 2964 (CH-aliph.); 2917; 2865; 1610; 1579; 1541; 1512; 1488; 1303; 1152; 1077; 816.
H-NMR (250 MHz, THF-d8): 1,38 (s, 9H, CH3-(tert.)-Butyl.); 2,34 (s, 6H, CH3-Tol.); 7,19 (m, 4H, CH-arom.); 7,87 (m, 4H, CH-arom.).
C-NMR(100 MHz, THF-d8): 21,0 (CH3-Tol.); 28,2 (CH3-(tert.)-Butyl.); 36,9 (C(quart.), tert.-Butyl.); 124,6 (br., CH-arom.); 129,8 (CH-arom.); 136,4 (ipso-Me.); 141,2 (C(quart.), ipso-N); 165,2 (C(quart.), C4/C5-Imidazol) 204,1 (C2-Imidazol).
UV-VIS(CHCl3): 295(4,1); 460(3,7); 604(2,9).
2-(tert.)-Butyl-5-(4-(tert.)-Butylphenylamino)-4-(4-(tert.)-Butylphenylimino)-4H-imidazol 3b:
Ausbeute: 62 %, orange Kristalle, m.p.: 75-79°C.
MS(CI): 417(100)[M+1]; 401(10); 359(45).
IR(ATR): 3346 (NH); 3072 (CH-arom.); 2961 (CH-aliph.); 2901; 2869; 1620; 1578; 1543; 1305; 1488; 1265; 1159; 1075; 839; 665; 618.
H-NMR (400 MHz, CDCl3): 1,33 (s, 18H, CH3-(tert.)-Butylphen.); 1,41 (s, 9H, CH3-(tert.)-Butyl); 7,44 (m, 4H, CH-arom.); 7,85 (m, 4H, CH-arom.).
C-NMR(100 MHz, CDCl3): 27,9 (CH3-(tert.)-Butyl.-imidazol); 31,3(CH3-(tert.)- Butylphen.) 34,6 (C(quart.), tert.-Butylphen.); 123,3 (br., CH-arom.); 126,1 (CH-arom.); 139,3 (ipso-N); 149,6 (C(quart.), ipsoAlk); 163,4 (C(quart.), C4/C5-Imidazol); 204,4 (C2-Imidazol).
UV-VIS(CHCl3): 297(4,2); 461(4,2).
1-(5-(4-(tert.)-Butylphenylamino)-4-(4-(tert.)-Butylphenylimino)-4H-imidazol-2-yl)-adamantan 3c:
Ausbeute: 40 %, orange Kristalle, m.p.: 198-201° C.
MS(CI): 495(100)[M+1]; 437(30); 353(40); 135(13); 93(37); 83(30); 71(41).
IR(ATR): 3338 (NH); 3069 (CH-arom.); 3032; 2963 (CH-aliph.); 2901; 2852; 1619; 1610; 1578; 1543; 1453; 1326; 1281; 1056; 840; 673.
H-NMR (400 MHz, CDCl3): 1,32 (s, 18H, CH3-(tert.)-Butyl.); 1,80 (s, 6H, CH2); 2,10 (s, 9H, CH/CH2); 7,40 (m, 4H, CH-arom.); 7,77 (m, 4H, CH-arom.).
C-NMR(100 MHz, CDCl3): 28,0 (CH2); 31,4(CH3-(tert.)-Butyl.); 34,6 (C(quart.), tert.-Butyl.); 36,8 (CH2); 38,6 (C(quart.), ipso-Adam.); 39,5 (CH); 123,2 (br., CH-arom.); 126,0 (CH-arom.); 138,7 (br., ipso-(tert.)-Butyl.); 149,5 (C(quart.), ipso-N); 163,7 (C(quart.), C4/C5- Imidazol) 203,3 (C2-Imidazol).
UV-VIS(CHCl3): 302(4,1); 462(4,0); 559(3,2); 604(3,2).